Site Map Sitemap Addiction Aging & Geriatrics Allergies Alzheimer's Anemia Anesthesiology & Pain Management Animal Research Artificial Intelligence (AI) Asthma Autism Autoimmune Conditions Awards & Honors Biochemistry Bioengineering Blood Cancers Blood disorders (Hematology) Breast Cancer C Diff Cancer Cardiology Celiac Disease Cellular & Molecular Biology Cervical Cancer Chronic Fatigue Syndrome Cold & Flu Colorectal Cancer Community Programs COPD COVID-19 Data Sciences Depression Dermatology Diabetes Digitally Driven Diversity, Equity & Inclusion Drug Development Ear, Nose & Throat Emergency Medicine Environment & Sustainability Epidemiology & Population Health Ethics Gastroenterology & Hepatology Genetics Gestational Diabetes Global Health Health Equity Health Policy Hepatitis HIV, AIDS HPV (Human Papillomavirus) Huntington's Disease Immunology Immunotherapy Infectious Diseases Inflammatory Bowel Syndrome Innovation & Technology Internal Medicine Kidney Health (Nephrology) Liver Cancer Lung Cancer Lung Health Malaria Maternal Health Medical Education Medical Research Men's Health Microbiology Multiple sclerosis Neurobiology Neurology & Neurosurgery Nutrition Obstetrics & Gynecology Ophthalmology Orthopaedics Osteoporosis Ovarian Cancer Palliative Care Pancreatic Cancer Parkinson's Pathology Patient Care Pediatrics Physiology Precision Health Preventive Medicine Primary Care Prostate Cancer Psychiatry & Mental Health Radiology Rheumatoid Arthritis RSV (Respiratory Syncytial Virus) Salmonella SHC Tri-Valley Skin Cancers Sleep SM Partners Sports Medicine Stanford Health Care Stanford Medicine Stanford Medicine Children's Health Stanford School of Medicine Stem cells Stroke Surgery Transplantation Tuberculosis Type 1 Diabetes Type 2 Diabetes Undiagnosed Uniquely Stanford Urology Vaccines VF Wellness Women's Health Latest Articles How a cultural exchange from Palo Alto to rural India is advancing perinatal health Author Laura HedliPublished on April 25, 2024May 3, 2024 Ask Me Anything: Everything to know about allergy season — and more Author Hanae ArmitagePublished on April 25, 2024April 25, 2024 Inequity of genetic screening: DNA tests fail non-white families more often Author Erin DigitalePublished on April 18, 2024April 22, 2024
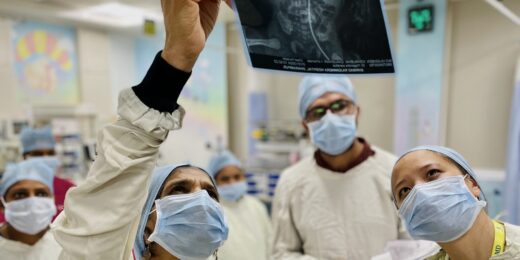
How a cultural exchange from Palo Alto to rural India is advancing perinatal health Author Laura HedliPublished on April 25, 2024May 3, 2024
Ask Me Anything: Everything to know about allergy season — and more Author Hanae ArmitagePublished on April 25, 2024April 25, 2024
Inequity of genetic screening: DNA tests fail non-white families more often Author Erin DigitalePublished on April 18, 2024April 22, 2024